Klinische Therapie mit Genschere: Mutationen einfach wegschnippeln
In der EU ist jetzt die erste klinische Therapie mit einer Genschere zugelassen. Welche Chancen ergeben sich daraus für Patienten mit Erbkrankheiten?
Wenn Eizelle und Spermium fusionieren, ist der Bausatz für einen neuen Menschen komplett. Damit dabei nichts schief geht, braucht es eine detaillierte Bauanleitung. Diese ist als Erbinformation in Form von Genen auf der DNA gespeichert. Im Falle von Erbkrankheiten ist es aber so, dass Neugeborene mit einem Bauplan ins Leben starten, der gesundheitliche Schwierigkeiten mit sich bringt.
Sichelzellanämie und Thalassämie sind die zwei häufigsten Erbkrankheiten der Menschheit. Beide Leiden zählen zu den sogenannten Hämoglobinopathien. Das bedeutet, dass Mutationen im Gen, das den Bauplan für das Eiweiß Hämoglobin enthält, die Ursache für die Erbkrankheit sind.
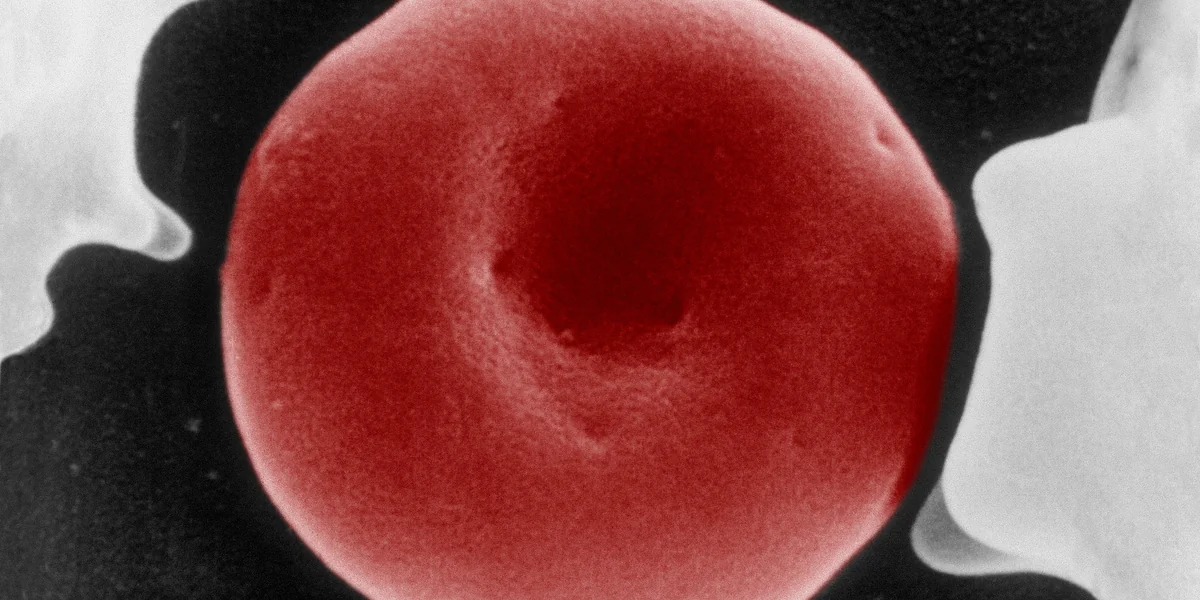
Als roter Blutfarbstoff sorgt Hämoglobin im Inneren jedes roten Blutkörperchens dafür, dass Sauerstoff in der Lunge aufgenommen und im Gewebe wieder abgegeben werden kann. Bei Patient:innen mit Hämoglobin-Störungen bereitet genau dieser Sauerstofftransport durch die Blutbahn massive Probleme.
Im Falle der Sichelzellerkrankung ist es etwa so, dass rote Blutkörperchen, die normalerweise wie ein Donut mit einer dünnen Membran in der Mitte aussehen, halbmondförmig sind. Diese Deformierung führt dazu, dass die roten Blutkörperchen weniger biegsam sind und dazu neigen, in kleinen Gefäßen zu verklumpen und Entzündungen auszulösen. Die Folge sind Infarkte in allen Organen, heftige Schmerzepisoden und Infektanfälligkeit. Ohne Behandlung lassen sich bereits ab dem ersten Lebensjahr Organschäden feststellen.
In Ländern, in denen Sichelzellerkrankte schlecht oder gar nicht versorgt werden, sterben viele sehr früh – vor allem an Infektionen und schweren Sichelzellkrisen, bei denen es zu mehreren Gefäßverschlüssen gleichzeitig kommt.
Patient:innen mit einer schweren Thalassämie können überhaupt kein Hämoglobin bilden und sind ihr Leben lang transfusionsbedürftig, was ebenfalls zu lebensbedrohlichen Organschäden führt. Beide Erkrankungen sind in Teilen des Mittelmeerraums sowie vom Nahen Osten bis nach Südostasien verbreitet. Die Sichelzellerkrankung tritt jedoch auch in Afrika und Nordeuropa auf.
Bisher gab es nur eine Behandlungsmethode
Hämoglobinopathien ließen sich bislang nur durch Stammzelltransplantationen heilen. Dabei werden körpereigene Blutstammzellen durch eine Chemotherapie entfernt und anschließend die gesunden Stammzellen eines geeigneten Spendenden transplantiert. Anfang Februar hat die EU-Kommission nun eine Gentherapie auf Basis der Genschere Crispr/Cas zugelassen.
Im Rahmen der Zulassungsstudie ist die allererste Patientin am Universitätsklinikum Regensburg behandelt worden, einer der weltweit erfahrensten Einrichtungen für Hämoglobinopathien. Selim Corbacioglu, Leiter der Abteilung für Pädiatrische Hämatologie, Onkologie und Stammzelltransplantation, hat diese allererste Genscheren-Therapie 2019 durchgeführt. Er berichtet, dass die heute 25-jährige Patientin seitdem gesund ist und keine Bluttransfusionen mehr benötigt.
Das Verfahren funktioniert so: Nach der Entnahme patienteneigener Stammzellen werden diese im Labor unter Strom gesetzt und die Zellwände dadurch durchlässig. So gelangt die Genschere ins Zellinnere und schneidet die DNA an einer vorgegebenen Stelle. An dieser Schnittstelle können einzelne DNA-Bausteine eingefügt, entfernt oder modifiziert werden.
Bei dem exa-cel-Verfahren wird eine Sequenz im Genom zerstört, die nach der Geburt die Produktion von sogenanntem fetalem Hämoglobin ausschaltet. Es bindet Sauerstoff so effizient, dass Ungeborene diesen buchstäblich aus der Plazenta heraussaugen. Nach der Geburt wird das fetale Hämoglobin nicht mehr benötigt und die Gensequenz dafür durch einen „DNA-Schalter“ ausgeknipst.
Corbacioglu sagt: „Mit der Genschere machen wir diesen Schalter kaputt.“ Die Technologie heilt Sichelzell-Erkrankte also indirekt: Nach der Transplantation der veränderten Stammzellen wird zwar immer noch fehlerhaftes Hämoglobin, gleichzeitig aber auch gesundes, fetales Hämoglobin vom Körper hergestellt. Beide Varianten sorgen zusammen dafür, dass rote Blutkörperchen ihre normale Funktion zurückgewinnen und der Sauerstofftransport durch die Gefäße ohne Probleme gelingt.
Noch weiß man nichts über die Langzeitfolgen
„Das Tolle an der Gentherapie ist, dass wir damit allen Patienten mit einer Hämoglobinopathie, für die es keinen passenden Stammzellspender gibt, eine heilende Therapie anbieten können“, sagt der Hämatologe. Als Allheilmittel betrachtet er die Innovation aber nicht. Exa-cel sei eine hervorragende zusätzliche Therapieoption, werde aber voraussichtlich mehrere Millionen Euro pro Behandlung kosten, während für Stammzelltransplantationen nur ein Bruchteil davon anfällt – bei vergleichbarem Ergebnis.
Außerdem sei die Datenlage noch dünn: „Wir führen seit 50 Jahren Stammzelltransplantationen durch und wissen genau, welche Nebenwirkungen und Spätfolgen es gibt. Bei der Gentherapie wissen wir hingegen nichts über das Risiko für spätere Krebserkrankungen, die Lebenszeit genetisch veränderter Zellen oder langfristige Konsequenzen des ‚neuen‘ Hämoglobins für Patienten.“ Trotz allem bleibt exa-cel ein wissenschaftlicher Durchbruch. Denn es handelt sich um den ersten klinischen Einsatz der Genschere überhaupt.
Es gibt verschiedene Ansätze für Gentherapien. Welcher davon am besten wirkt, hänge vor allem vom therapeutischen Zielorgan ab, sagt Corbacioglu. Zellen können entweder dem Körper entnommen und dann genetisch verändert werden, wie bei der Sichelzellanämie, oder das Gentherapeutikum wird in einem inaktivierten Virus verpackt, der als Taxi zum erkrankten Organ dient.
So ein Viren-Taxi gibt es etwa für Menschen mit Hämophilie, auch Bluterkrankheit genannt. Es steuert Leberzellen an und liefert das Gen für den fehlenden Gerinnungsfaktor ab, das dann zigfach an verschiedenen Stellen zufällig in die DNA eingebaut wird.
Corbacioglu: „Bei Blutzellen ist der Vorteil, dass man sie dem Körper entnehmen kann. Bei Hämophilie ist das anders. Wir können nicht einfach die Leber rausholen und ihre Zellen behandeln.“ Ein weiterer Unterschied ist, dass bei der Variante mit dem Viren-Taxi das Genom mehr als 100.000 Mal auseinandergebrochen wird, um das ‚neue‘ Gen einzubauen. „Mit Crispr/Cas können wir aber sehr genau bestimmen, wo im Genom die gewünschte Veränderung stattfindet. Das ist ein großer Vorteil.“
Außerdem sei es billiger, ohne Viren zu arbeiten, und die Crispr/Cas-Methode deshalb zukunftsträchtiger. Corbacioglu ist überzeugt, dass es innerhalb der nächsten 15 Jahre möglich sein wird, eine Crispr/Cas-basierte Therapie auch im Körper durchzuführen.
Gemeinsam für freie Presse
Als Genossenschaft gehören wir unseren Leser:innen. Und unser Journalismus ist nicht nur 100 % konzernfrei, sondern auch kostenfrei zugänglich. Alle Artikel stellen wir frei zur Verfügung, ohne Paywall. Gerade in diesen Zeiten müssen Einordnungen und Informationen allen zugänglich sein. Unsere Leser:innen müssen nichts bezahlen, wissen aber, dass kritischer, unabhängiger Journalismus nicht aus dem Nichts entsteht. Dafür sind wir sehr dankbar. Damit wir auch morgen noch unseren Journalismus machen können, brauchen wir mehr Unterstützung. Unser nächstes Ziel: 50.000 – und mit Ihrer Beteiligung können wir es schaffen. Setzen Sie ein Zeichen für die taz und für die Zukunft unseres Journalismus. Mit nur 5,- Euro sind Sie dabei! Jetzt unterstützen





meistkommentiert